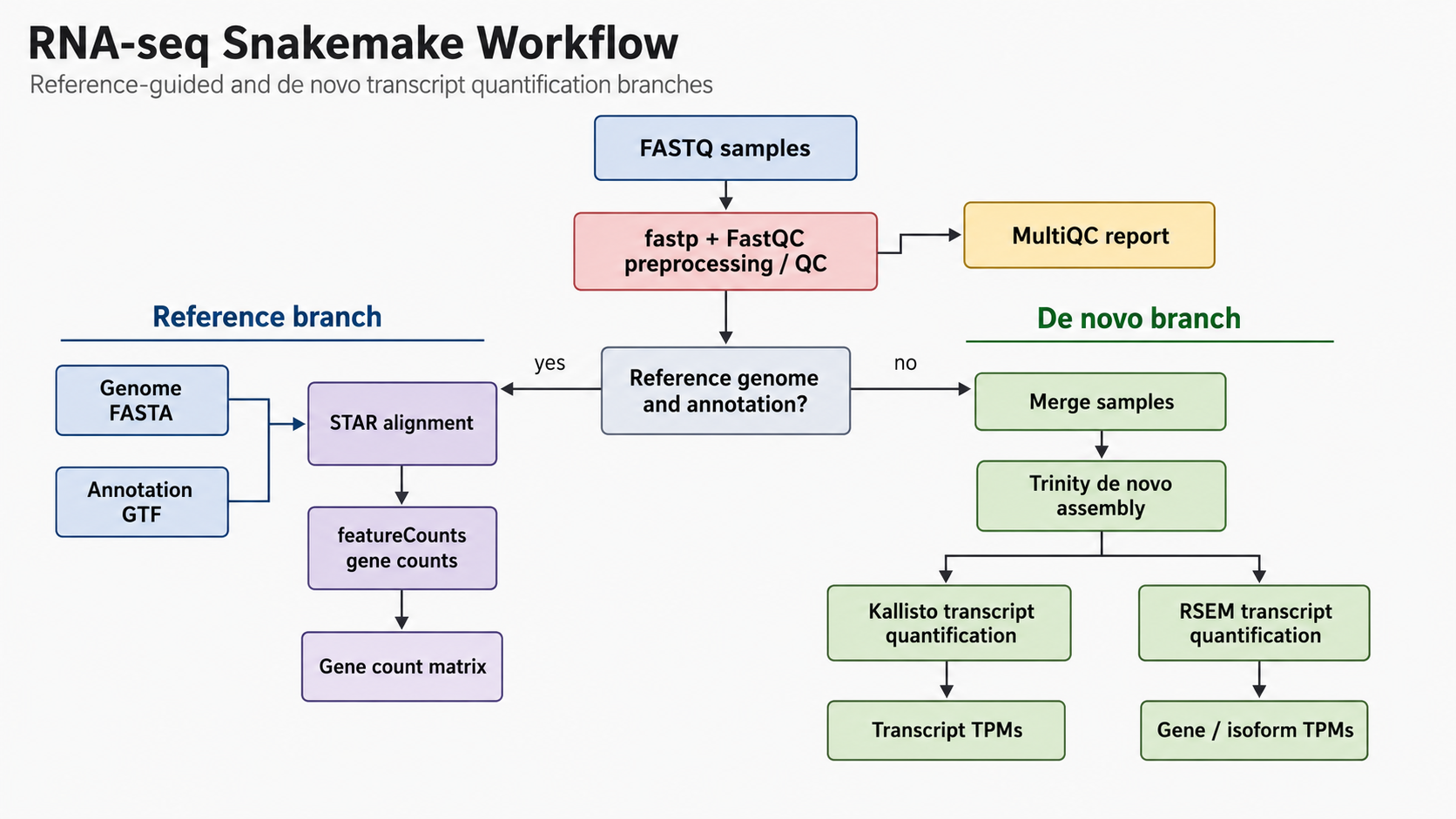
RNA-seq Transcript Quantification Workflow
Built a reproducible RNA-seq workflow in Snakemake with reference-guided analysis using STAR and featureCounts, plus de novo transcript quantification with Trinity, Kallisto, and RSEM.
Work
Reproducible workflows for genomics, metagenomics, transcript quantification, and biomedical data analysis.
Built a reproducible RNA-seq workflow in Snakemake with reference-guided analysis using STAR and featureCounts, plus de novo transcript quantification with Trinity, Kallisto, and RSEM.
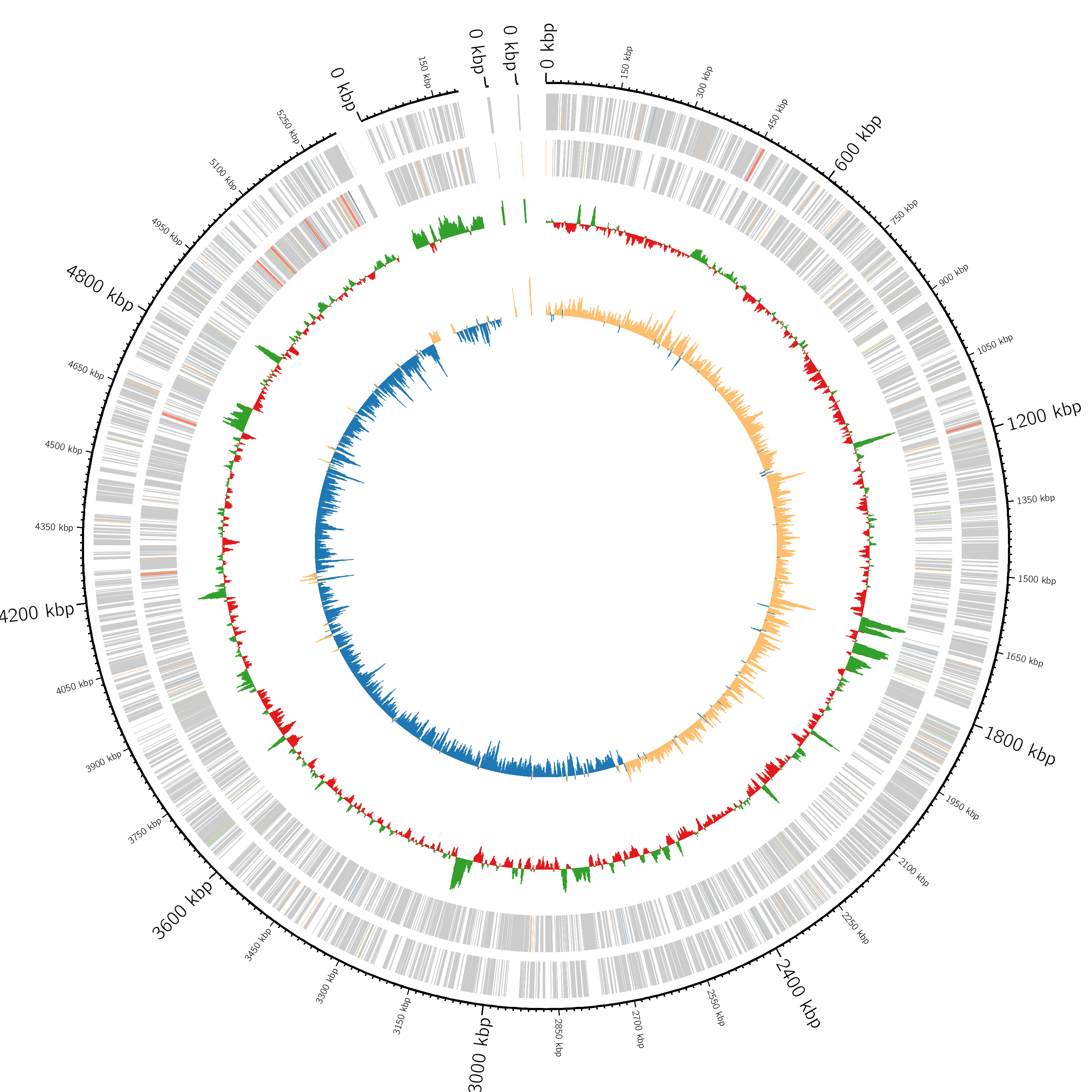
Built a Snakemake workflow for bacterial whole-genome sequencing analysis, including QC, short-read or hybrid assembly, polishing, annotation, core-genome phylogeny, and optional MLST, AMR, virulence, and plasmid screening.
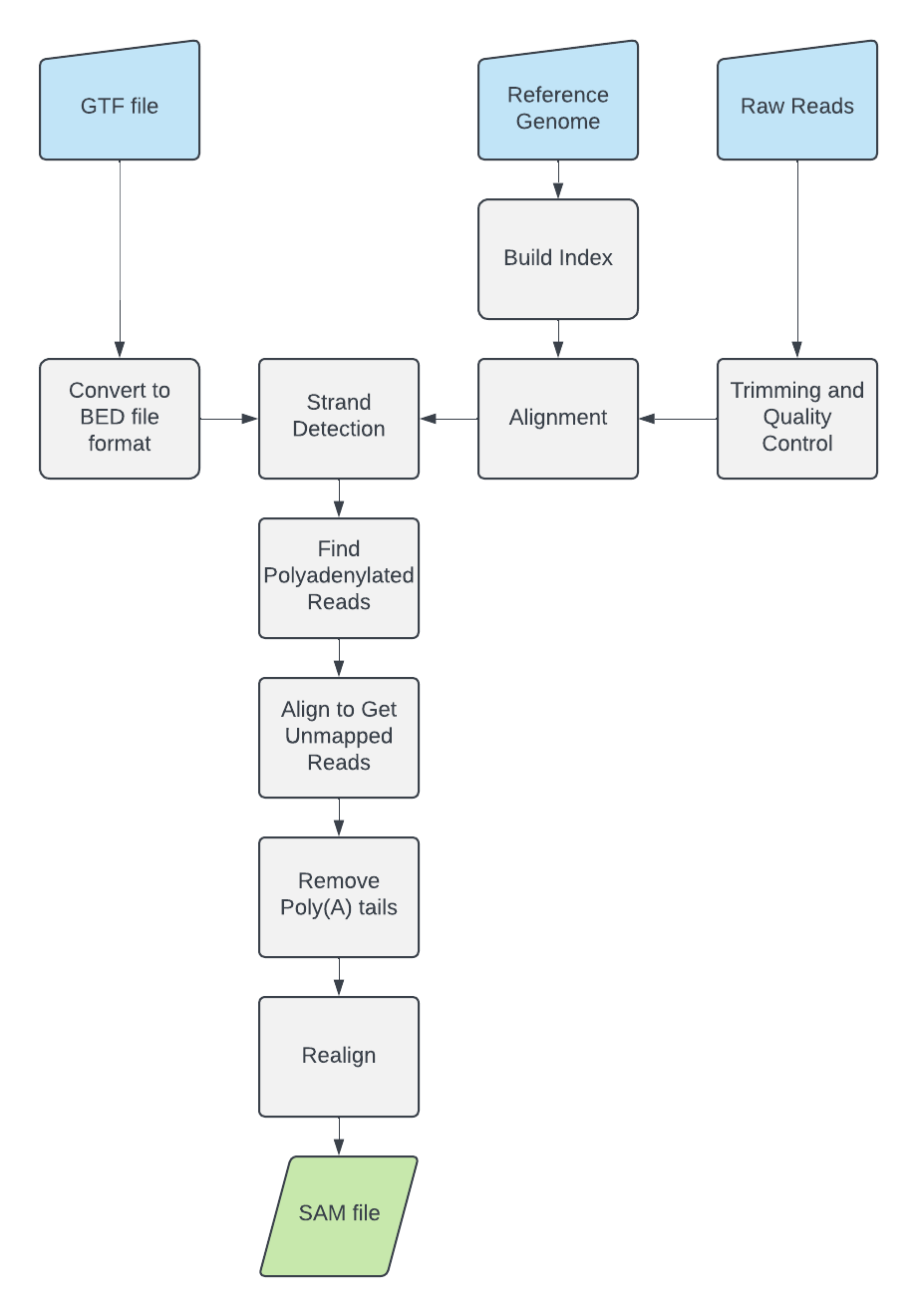
Designed and implemented a Nextflow DSL2 workflow to recover RNA-seq reads with visible poly(A)/poly(T) tail signal, trim the tail sequence, and realign the mRNA-derived read body to a reference genome.
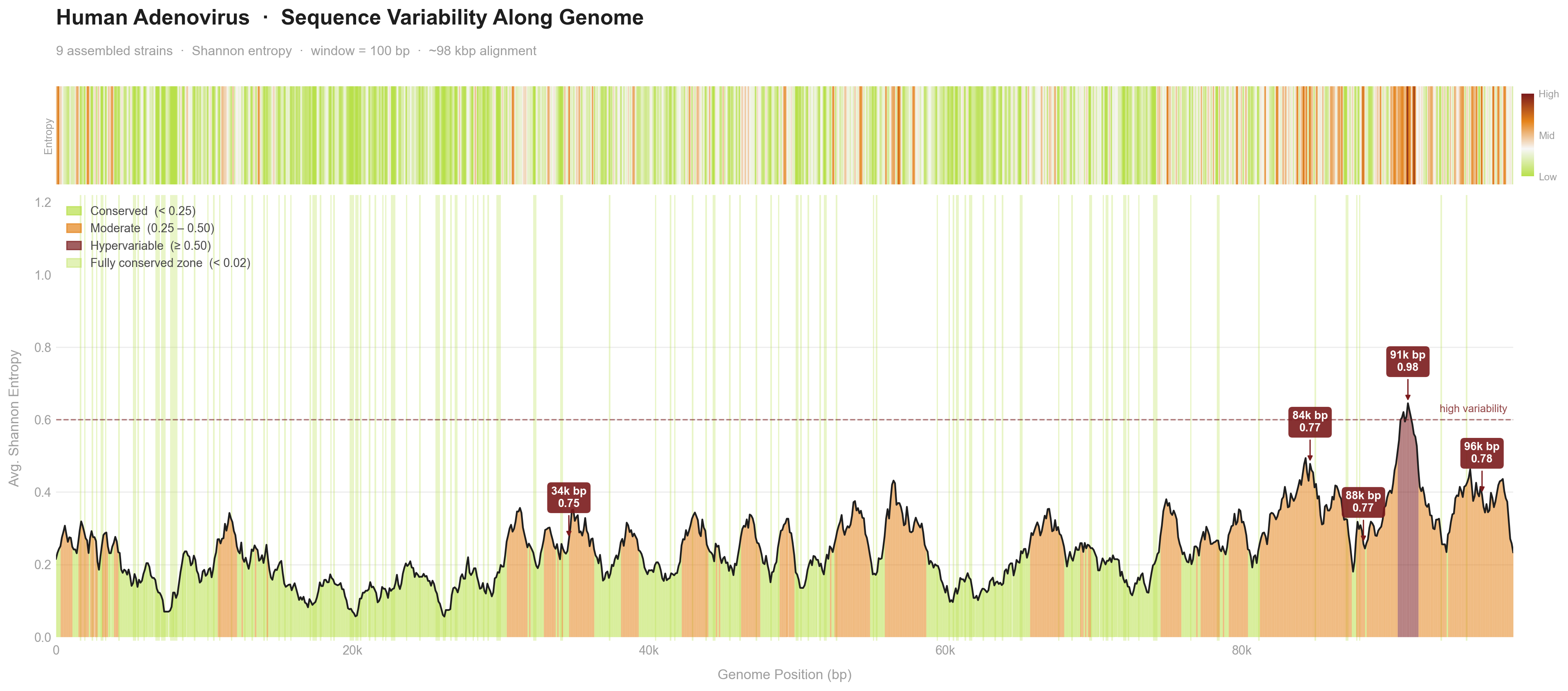
Built a Snakemake pipeline for Human Adenovirus genome assembly, comparative genomics, and phylogenetic analysis, from raw paired-end reads through QC, assembly, scaffolding, consensus calling, variability plots, and tree generation.